In our second research project, we investigate middle and late 3d transition metal complexes in bulky bis(alkoxide) ligand environments as vehicles for nitrene and carbene transfer. Alkoxides are weak-field ligands, and bulky alkoxides further increase the electrophilicity of the metal center by creating a low-coordinate complex. This coordination environment is expected to produce reactive nitrene and carbene complexes. Several bulky alkoxide ligands (monodentate [OCtBu2Ph] and [OCtBu2(3,5-Ph2C6H3)] and related chelating bis(alkoxide)s) have been synthesized, and their Cr(II)-Co(II) bis(alkoxide) complexes [M(OR)2(THF)2] obtained and characterized. As an indication of the steric hindrance of the alkoxide ligands, the complexes generally exhibit distorted seesaw geometry, with wide RO-M-OR (>140 °) and narrow THF-M-THF angles.
2(THF)2.jpg)
![Fe[OO]Ph(THF)2.jpg](https://static.wixstatic.com/media/594d5b_425256264bac478b9080828f705d474d~mv2.jpg/v1/fill/w_178,h_140,al_c,q_80,usm_0.66_1.00_0.01,enc_avif,quality_auto/Fe%5BOO%5DPh(THF)2.jpg)
2(THF)2.jpg)
We have shown that bulky bis(alkoxide) ligation has a profound effect on the reactivity of the metal center. Thus, an addition of an oxidizable ligand/group (azide, diazoalkane, ylide) to [M(OR)2] precursor (M = Fe, Co) generally leads to the formation of [M(OR)2(X•)] species, which can exhibit rare or unprecedented reactivity. Treatment of Fe(OR)2(THF)2 with aliphatic azides leads to the reductive coupling of azides to form iron(III) hexazene complex [(RO)2Fe(μ-κ2:κ2-RNNNNNNR)Fe(OR)2]. In contrast, the reaction of [Fe(OR)2] with aromatic azides forms highly reactive [M(OR)2(NAr)] species that catalyzes nitrene homo- and heterocoupling to yield azoarenes (ArN=NAr). Several different catalysts were developed allowing formation of symmetric and asymmetric azoarenes featuring both bulky (e.g. mesityl) or non-bulky (phenyl) Ar groups.


Our current chemistry in the iron bis(alkoxide) project focuses on the synthesis and reactivity of iron-carbene compexes. Our initial attempts to obtain iron carbenes involved diazo precursors. However, treatment of Fe(OR)2(THF)2 with diazo precursors results in rare reductive coupling through the terminal nitrogens, similar to the reductive coupling of aliphatic azides. The product of coupling is sufficienly stable to be isolated and structurally characterized for diazoester precursors. We have also described an “inverted” or remote carbene radical that originated from the reaction of [Fe(OR)2] with iodonium ylides, and its efficient carbene transfer reactivity to olefins. Our current research in this project explores the generality of this motif, and additional metal-mediated carbene transfer reactions.


For Co, we have reported the synthesis and characterization of the first stable high-valent alkylidene-like cobalt-carbene Co(OR)2(=CPh2). This compound was obtained by the reaction between Co(OR)2(THF)2 and diphenyldiazomethane, and was accompanied by N2 release. The electronic structure analysis of this low-spin compound suggested a compound intermediate between Co(III)-carbene radical and a genuine Co(IV)-alkylidene. In contrast, one-electron reduced product (CoCp*2)[Co(OR)2(CPh2)] is a high-spin Co(II)-carbene radical. [Co(OR)2] was found to catalyze efficient carbene transfer to isocyanides CNR' to form ketenimines R2C=C=NR'. Our current work in this project targets additional Co(OR)2-carbene species in different oxidation states, and their carbene transfer reactivity.


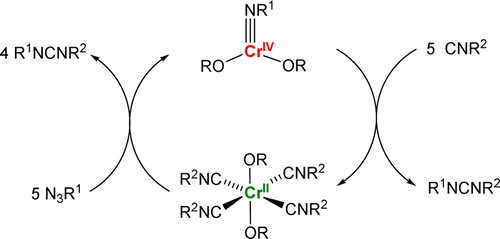
We have also investigated the synthesis and reactivity of low-coordinate [Cr(OR)2] complexes. Low-coordinate Cr(OR)2(NR’) imido complexes were found to catalyze nitrene transfer to isocyanides, forming carbodiimides in good yields. Additionally, [Cr(OR)2] precursors were able to catalyze reductive (pinacol) coupling of aldehydes, affording Cr(IV)-diolate products. In contrast, CO2 inserted into the Cr-OR bond to afford dichromium(II) tetracarboxylate. Our current research efforts in this project focus on the synthesis and reactivity of Cr-carbenes.